
Overview
 The goal of OPL’s proteomics research is to identify tumor-associated changes in the proteome using a mass spectrometry-based open screening strategy, and to translate this information – preferably insights relating to tumor biology – into protein-based clinical tests.
The goal of OPL’s proteomics research is to identify tumor-associated changes in the proteome using a mass spectrometry-based open screening strategy, and to translate this information – preferably insights relating to tumor biology – into protein-based clinical tests.
The activities of OPL are focused on 1) identifying protein markers for non-invasive (early) detection of cancer and for monitoring disease; 2) identifying new predictive markers/targets for (indiviualized) targeted therapy of cancer; and 3) developing/implementing innovative proteomics and data analysis strategies to enable the above.
Strategy
To enable large-scale protein identification and quantification, mass spectrometry is performed on high-resolution (tandem) instruments. Such platforms can not only be used to measure protein levels, but also to obtain information about primary protein structure (splice variants, isoforms, mutant variants, post-translational modifications such as phosphorylation). This type of information is crucial to gain molecular insight into the different states of cancer cells and their activation, to be able to develop drugs that specifically target implicated proteins, and select proteins to develop biomarker-related clinical applications.
For discovery experiments we use label-free shot-gun proteomics based on nanoLC-MS/MS and spectral counting or intensity-based quantitation. For large-scale validation and profiling studies we use data-independent mass spectrometry. Depending on the application, we may turn to antibody-based assays for final clinical applications.
References:
- Jimenez CR., Piersma SR., Pham TV. (2007) High-throughput and targeted in-depth mass spectrometry-based approaches for biofluid profiling and biomarker discovery. Biomark. Med. 1(4): 541-565. Review.
- Jimenez CR. (2008) Mass spectrometry-based proteomics: trends in tools and strategies. Eur. Pharm. Rev. 7 Apr 2008 (Industry Focus 2008): 24-25. Review.
- Rajcevic, U., Niclou, S., Jimenez, CR. (2009) Proteomics strategies for target identification and biomarker discovery in cancer. Fronti. Biosci. 14: 3293-3303. Review.
- Jimenez CR, Knol JC, Meijer GA, Fijneman RJ. (2010) Proteomics of colorectal cancer: overview of discovery studies and identification of commonly identified cancer-associated proteins and candidate CRC serum markers. J. Proteomics. 73(10):1873-1895. Review.
- Piersma SR, Labots M, Verheul HM, Jiménez CR. (2010) Strategies for kinome profiling in cancer and potential clinical applications: chemical proteomics and array-based methods. Anal. Bioanal. Chem. 397(8):3163-3171. Review.
- van Dijk KD, Teunissen CE, Drukarch B, Jimenez CR, Groenewegen HJ, Berendse HW, van de Berg WD. (2010) Diagnostic cerebrospinal fluid biomarkers for Parkinson’s disease: a pathogenetically based approach. Neurobiol. Dis. 39(3):229-241. Review.
- Jimenez CR, Verheul HM. Mass spectrometry-based proteomics: from cancer biology to protein biomarkers, drug targets, and clinical applications. Am Soc Clin Oncol Educ Book. 2014:e504-10.
- Lam SW, Jimenez CR, Boven E. Breast cancer classification by proteomic technologies: current state of knowledge. Cancer Treat Rev. 2014 Feb;40(1):129-38.
- Schaaij-Visser TB, de Wit M, Lam SW, Jiménez CR. The cancer secretome, current status and opportunities in the lung, breast and colorectal cancer context. Biochim Biophys Acta. 2013 Nov;1834(11):2242-58.
- de Wit M, Fijneman RJ, Verheul HM, Meijer GA, Jimenez CR. Proteomics in colorectal cancer translational research: biomarker discovery for clinical applications. Clin Biochem. 2013 Apr;46(6):466-79.
- Kranenburg O, Emmink BL, Knol J, van Houdt WJ, Rinkes IH, Jimenez CR. Proteomics in studying cancer stem cell biology. Expert Rev Proteomics. 2012 Jun;9(3):325-36.
- Pham TV, Piersma SR, Oudgenoeg G, Jimenez CR. Label-free mass spectrometry-based proteomics for biomarker discovery and validation. Expert Rev Mol Diagn. 2012 May;12(4):343-59.
- Bosch LJ, Carvalho B, Fijneman RJ, Jimenez CR, Pinedo HM, van Engeland M, Meijer GA. Molecular tests for colorectal cancer screening. Clin Colorectal Cancer. 2011 Mar 1;10(1):8-23.
- Jimenez CR, Knol JC, Meijer GA, Fijneman RJ. Proteomics of colorectal cancer: overview of discovery studies and identification of commonly identified cancer-associated proteins and candidate CRC serum markers. J Proteomics. 2010 Sep 10;73(10):1873-95.
Expertise of the OncoProteomics Laboratory
- Label Free (phospho)proteomics, shot-gun & data independent
- Global pSTY and pY specific phosphopeptide enrichment
- Analysis of various tumor tissue sample types (fresh-frozen, FFPE, RNA-left over fraction), biofluids, platelets and exosomes
- Bioinformatics, Statistics. Computational biology
Label Free (phospho)proteomics: shot-gun & data independent
Protein expression profiling
For protein expression profiling, we performed pioneering work in the area of label-free proteomics (Piersma et al., J Proteome Res. 2010; reviewed by Pham et al. in Expert Rev Mol Diagn. 2012) and improved the throughput of the GeLC workflow (Piersma et al., Proteome Sci. 2013).
Discovery proteomics for protein expression profiling typically makes use of our optimized “GeLC"-MS/MS workflow. This workflow allows for routine fractionation at the protein and peptide level, to enable in-depth analysis of complex biological and clinical samples at medium throughput (~8 hours of instrument time per sample).
In case sample complexity is low or intermediate, fractionation may be omitted and only short-stack electrophoresis is performed preparing a “blob” gel band for single shot nanoLC-MS/MS analysis (2 hours of instrument time per sample).
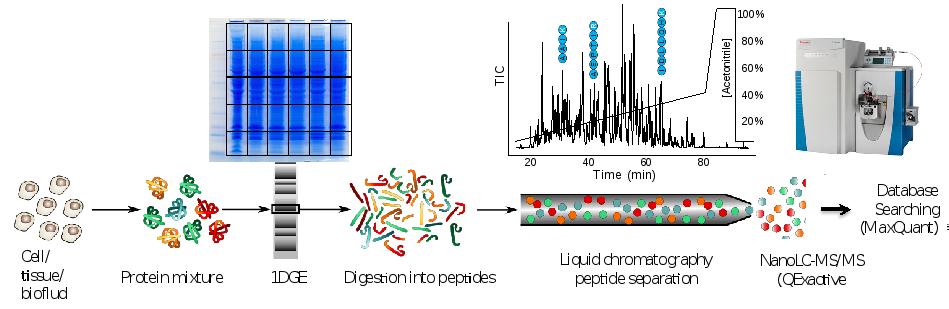
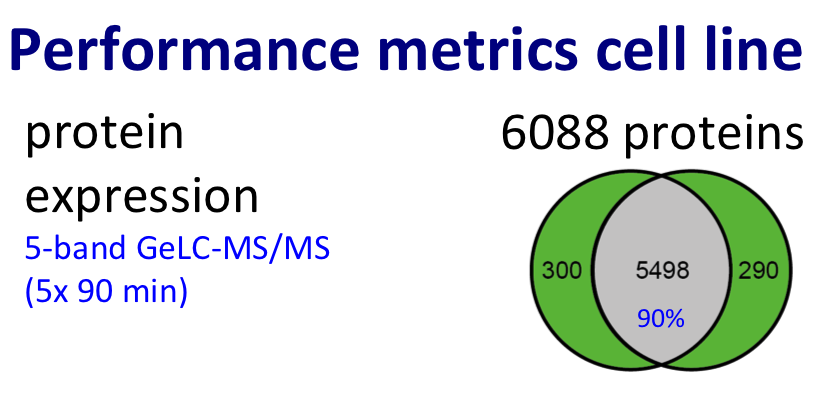
Figure. Proteomics workflow and performance metric cancer cell line.
Protein phosphorylation profiling
Global pSTY and pY specific phosphopeptide enrichment
For successful phosphoproteome analyses, phosphopeptides need to be enriched prior to analysis by nanoLC-MS/MS (~2 hours of instrument time per sample). The OPL has implemented and benchmarked two robust and reproducible modalities for phosphopeptide capture: (i) titanium oxide for global capture of phosphopeptides with pSer, pThr or pTyr residues, and (ii) a pTyr-specific antibody for selective capture of pTyr residues.
These workflows were bench-marked for label-free single shot global phosphoproteomics using titanium-oxide (Piersma et al., J Proteomics. 2015), and using different phosphotyrosine antibodies for phosphopeptide capture (Van der Mijn et al., J Proteomics. 2015).
In recent years, successful downscaling of these methods has enabled phosphoproteomic analyses of tumor biopsies in clinical studies (Labots et al., J Proteomics. 2017). Moreover, a dedicated integrative data analysis strategy has been developed to identify hyperactive phosphokinases in individual tumor samples. We foresee that this approach will be used in the future to complement genomic information in the setting of patient selection for targeted therapies and personalized medicine.
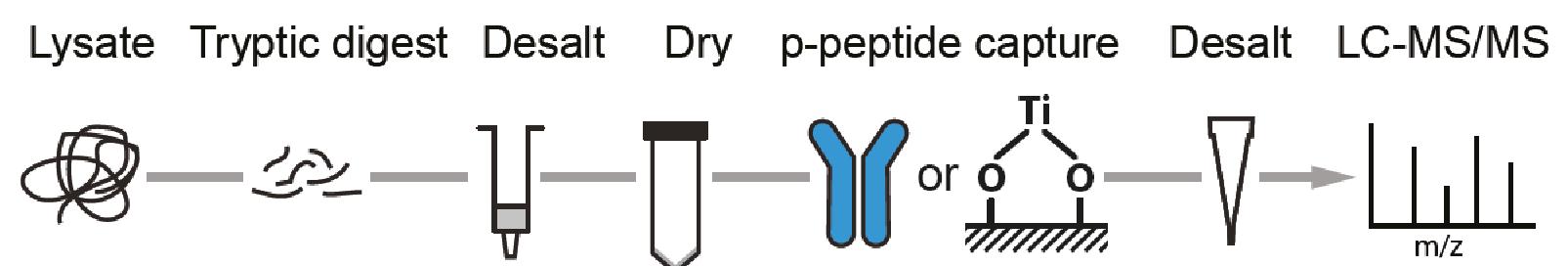
Figure. Phosphoproteomics workflow. Phosphopeptides are enriched using TiOx or a phosphotyrosine (pTyr)-specific antibody to enrich for the global or pTyr phosphoproteome respectively.
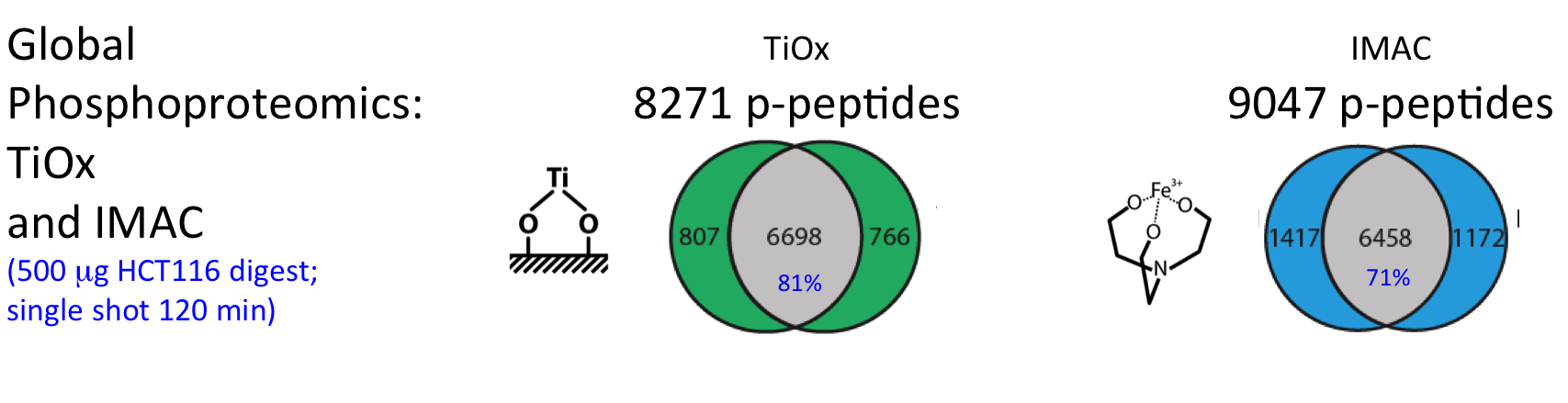

Figure. Performance phosphoproteomics workflows for cancer cell line
Data Independent Acquisition (DIA) MS
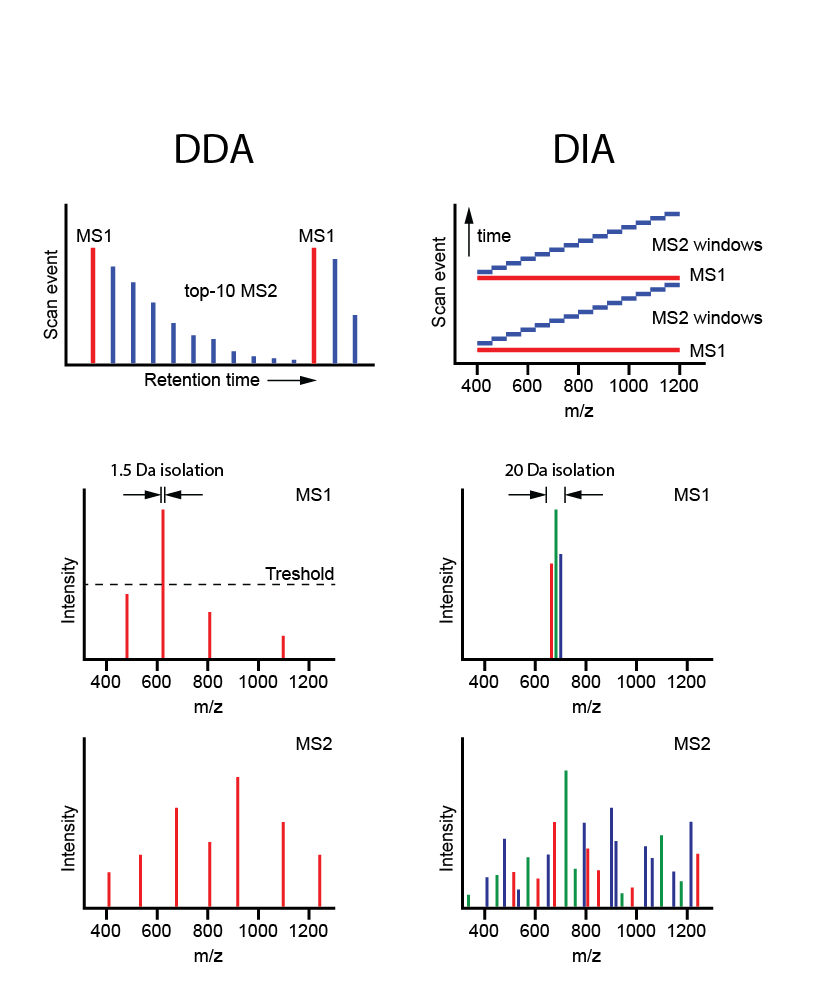 Figure 8. Data dependent acquisition (DDA< shot gun proteomics) vs. data independent acquisiton (DIA) MS[/caption]
Figure 8. Data dependent acquisition (DDA< shot gun proteomics) vs. data independent acquisiton (DIA) MS[/caption]
For large scale profiling of many samples (10s-100s), data-independent mass spectrometry is the MS approach of choice as it allows for in-depth analysis in only 2 hours of instrument time. This approach was recently developed by Aebersold et al., to get the best of discovery proteomics (in-depth protein inventories) and targeted MS (highly reproducible quantitation of selected proteins).
This revolutionary MS approach has been implemented at the OPL in 2017 and was made possible with funding for a new fast-scanning instrument from NWO-Middelgroot and the CCA.
More information on DIA-MS:
Whereas both global and targeted approaches entail the use of survey MS scans for final analysis in MS/MS mode (‘data-dependent acquisition’, DDA), in the novel DIA-MS strategy, consecutive mass windows are immediately subjected to MS/MS (data-independent acquisition, DIA), followed by targeted data extraction using a spectral library. The resulting ‘digital proteome maps’ (files of MS/MS data) can be (re-)analyzed and compared across multiple samples. Besides DIA-MS for large-scale tumor profiling, we are also exploring phospho-DIA-MS.
First results of our own hands show that DIA-MS has high precision quantitation (CVs < 5-6%) and low numbers of missing values, and therefore it may ultimately find its way to the clinical chemistry labs for routine diagnostics.
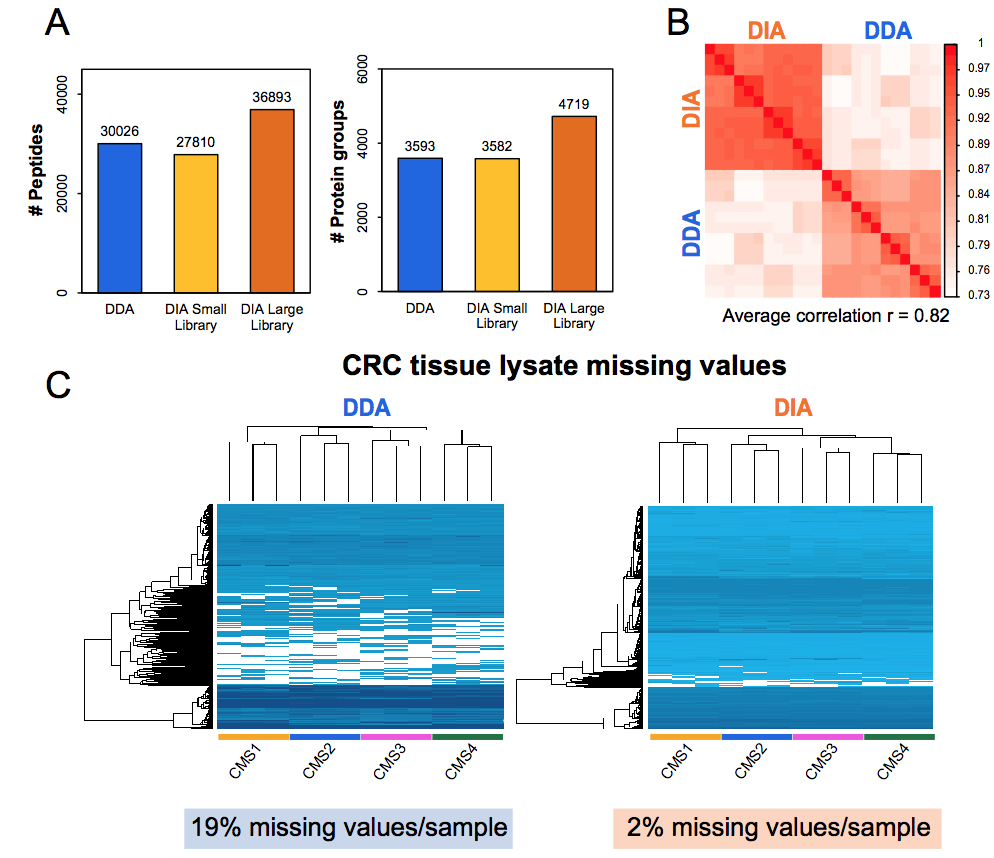
Figure. Performance DIA-MS versus shot-gun (DDA) workflows for cancer cell line (A) and tumor tissue (B-C)
Biological and clinical sample analysis
Cell and tissue analysis
The OPL has protocols for cell and tissue analysis, including fresh-frozen tissues, formalin-parafin embedded tissues, RNAbee left-over fraction and laser-capture micro-dissected tissue. For in-depth protein expression analysis of total tumor tissue lysates, 1-10 mg wet weight is enough (ie., biopsy level and smaller). If a large quantity is available (>50-100 mg), fractionation into tumor sub-proteomes/ sub-cellular fractions or protein complexes may enhance the sensitivity of detection of selected proteins of interest.
Extracellular proteomics
To obtain insight into the tumor microenvironment, and to develop non-invasive biomarker applications, workflows for the global analysis of secreted proteins (secretome), proximal biofluids (e.g., urine, sputum, cerebrospinal fluid and stool), as well as extracellular vesicles isolated from such matrices have been implemented and benchmarked at the OPL.
More specifically, protocols have been benchmarked and applied to the analysis of proteins on the cell surface, proteins that are secreted or released in the extracellular compartment (forming a ‘secretome’), proteins enclosed in extracellular vesicles (including ‘exosomes’) that are actively secreted by cells, and proteins in proximal (tumor-contacting) body fluids/excrements such as sputum and faeces.
Exosome proteomics: For highthroughput capture of exosomes, we recently benchmarked and optimized a novel high-throughput extracellular vesicle (enriched for exosomes) isolation protocol using cancer cell secretome (Knol et al, EuPA Open Proteomics 2016) and using urine (Bijnsdorp et al., J. Extracelular Vesicles 2017). Further implementation for of this promising highthroughput, clinically compatible EV capture method to cerebrospinal fluid (CSF) is on-going (Chiasserini in prep).
Biofluid/ CSF proteomics: Through our neuroproteomics projects, we obtained our expertise with biofluid proteomics, most notably CSF. To this end, we bench-marked two different abundant protein depletion filters for in-depth proteome analysis of CSF (Fratantoni et al., Proteomics. Clin. Appl. 2010).
Researchers may be trained in workflows of interest to prepare samples that can be subjected to in-depth discovery proteomics (as described above under protein expression profiling).
Dry lab: Bioinformatics, Statistics, Computational biology
Data analysis for biological interpretation
After protein identificatioin and quantification by sequence database searching using the freely web-based tool MaxQuant, for subsequent data interpretation, other web-based software (Cytoscape, String, Webgestalt) is used to perform functional data mining and network-based analyses. The latter involve visualization of protein-protein association networks which can aid in gaining insights into (the role of candidates in) tumor (cell) biology. Moreover, information gleaned from such networks may be used to prioritize candidate markers/targets for follow-up. Furthermore, for the analysis of phosphoproteomics data, OPL is actively engaged in the development of algorithms that enable prediction of hyperactive/driver kinases and visualize the data as kinase-substrate interaction networks (manuscript in prep).
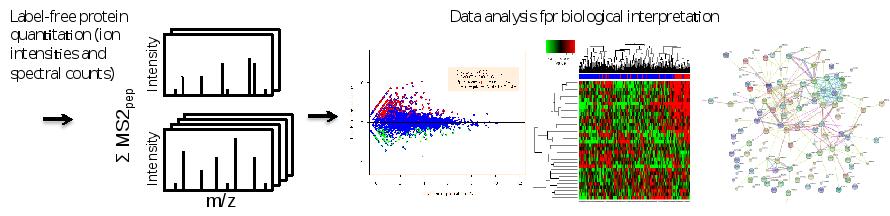
Figure. Data analysis workflow. Sequence database searching of combined MS and MS/MS data enables protein identification and quantification. Differential expression analysis and dedicated statistics can uncover proteins with altered abundance. Volcano plots, heat maps and protein interaction networks are complementary ways to get data overviews and biological information.
Proteogenomics
In the coming years, our research focus will include tumor-specific protein variants in pre-clinical and clinical samples (e.g., splice variants and mutated proteins) in order to develop specific protein markers. To allow for such research, OPL has set up a “proteogenomics” data analysis pipeline that can identify aberrant protein variants in cancer. In this approach, the MS/MS data are searched against genomics-based (sample/patient-specific) sequence databases that include disease variants. Moreover, in a KWF-funded project, we developed “Splicify”, a proteogenomic pipeline for identification of differentially expressed splice variants (Komor, Fijneman, Jimenez et al., MCP 2017).
OPL Research
Summary of Translational OncoProteomics research
Below, a summary is given of OPL research activities and multi-disciplinary collaborations involving different tumor types in each research line, with listing of publications of the last 3 years and the on-going projects with Jimenez as PI or coPI. In the appendix 1 of the OPL progress report 2015-2017, abstracts can be found of each research project.
For an impression of OPL research, click here to see video “A Tuesday afternoon at the VUmc OncoProteomics Laboratory” https://quik.gopro.com/v/cW3OzFSlO3/
1. Identification of (phospho)protein markers/targets for tumor subtyping, targeted treatment of cancer and tailored therapy for individual patients.
In 2014, research was started to uncover protein biomarkers for CRC subtype classification linked to prognosis (collaboration with Prof. Dr. Jan Paul Medema (AMC) and Prof.Jan Ijzermans (EMC) and partners KWF Alpesd’hu6 Connection consortium). CRC subtype markers have been identified in a set of 40 tumors and 25 cell lines and validated in an independent series of CRCs from Rotterdam as well as in external TCGA CRC tumor proteome data (Nature 2014). Currently a DIA-MS-based protein classifier is developed for profiling a third CRC biopsy series to be obtained in a clinical trial that aims to link CRC subtypes to (lack of) response to chemotherapy.
Large scale protein profiling is also done on a FFPE series of lung cancers (NSCLC) of patients that received adjuvant platina-based therapy, with the goal of identifying biomarkers for cisplatin response prediction (KWF project with Sjaak Burgers and Egbert Smit, NKI).
Protein phosphorylation by kinases plays a key role in the crucial change of the activity and/or wiring of intracellular signaling pathways and in the action of machinery that transforms cells into cancer cells. Recent analysie of AML cell lines and patient blasts (collaboration Dr. Jeroen Janssen, Dr. Jacqueline Cloos), colorectal adenomas and carcinomas (collaboration Beatriz Carvalho, Gerrit Meijer, NKI), patient-derived xenograft (PDX) models of colorectal cancer (collaboration with Prof. Livio Trusolini, IRCCS, Italy) and pancreatic cancer (collaboration dr. Elisa Giovanetti, Dr. Maarten Bijlsma), have shown the power of phosphoproteomics to uncover hyperactive kinases and identify known and novel “driver kinases” that are thought to play a central role in oncological transformation/progression. Phosphoproteomics of PDX models is also successfully being applied in the context of Homologous Recombination Deficiency in triple negative breast cancer (KWF project with Jos Jonkers).
Successful downscaling of the phosphotyrosine workflow and application to tumor biopsies has uncovered drug-specific signatures in a clinical trial (collaboration Mariette Labots and Henk Verheul). Currently, tumor needle biopsies are being collected in multiple clinical trials of the Dept. Medical Oncology (IMPACT-CRC, Soprano, Reposit) of the national Center for Personalized Cancer Treatment. In the coming years, we aim to perform phosphoproteomics of these tumor biopsies to establish its value for patient selection for targeted therapy.
Publications
- Johannes C. van der Mijn, Henk J. Broxterman, Jaco C. Knol, Sander R. Piersma, Richard R. De Haas, Henk Dekker, Thang V. Pham, Victor W. Van Beusechem, Balazs Halmos, James W. Mier, Connie R. Jiménez*, Henk M.W. Verheul*. Sunitinib activates Axl signaling in renal cell cancer. Int. J. Cancer 2016; *Shared senior authors; Accepted manuscript
- Le Large TYS, Bijlsma MF, Kazemier G, van Laarhoven HWM, Giovannetti E, Jimenez CR. Key biological processes driving metastatic spread of pancreatic cancer as identified by multi-omics studies. Semin Cancer Biol. 2017 Mar 30. pii:S1044-579X(17)30066-4.
On-going projects
- Discovery and clinical validation of novel protein biomarkers for homologous recombination deficient breast cancer (KWF VU2013-6020 Dutch Cancer Society, PIs Jimenez, Jonkers (NKI), Van Diest (UMCU))
- Tumor-specific protein biomarkers for early detection of colorectal cancer (KWF NKI2014-6025, PIs Fijneman (NKI), Jimenez, Meijer (NKI))
- The Molecular Signalling Pathways of Folliculin (FLCN): a Tumor Suppressor in Birt-Hogg Dubé Hereditary Kidney Cancer (VUmc PhD student Dept. Clin. Genetics with Wolthuis).
- Improving clinical management of colon cancer through CONNECTION, a nation-wide Colon Cancer Registry and Stratification effort. (KWF/Alpe d’Huzes UvA2013-6331, PIs Medema (AMC), Van Krieken (RUMC), IJzermans (EMC), Jimenez, Koopman (UMCU)).
- Response prediction for cisplatin-based treatment regimens in non-small cell lung cancer using a protein-based assay (KWF VU2014-6816, PIs Jimenez (VUmc), Grunberg (VUmc/RUMC), Burgers (NKI))
- Phosphoproteomics for therapy response prediction in solid tumors with a focus on colorectal cancer (Vitromics-sponsored PhD project, PIs Verheul and Jimenez)
- AML phosphoproteomics for personalized therapy (CCA-sponsored PhD project, VUmc PIs Janssen, Jimenez, Verheul)
- Signaling pathways involved in colorectal adenoma-to-carcinoma progression (KWF NKI2014-6813 Dutch Cancer Society, PIs Carvalho (NKI), Jimenez, Meijer (NKI))
- Phosphoproteomics for therapy response prediction in pancreatic cancer (VUmc-AMC alliance project 2014, PIs Giovanetti (VUmc), Jimenez, Bijlsma (AMC))
- How KRAS mutations in colon cells are linked to a new cancer hallmark: cohesion weakness (CCA 2015, PIs De Lange, Jimenez, Wolthuis)
- Interrogating the phosphoproteome for targets and markers in pancreatic cancer (KWF VU2016-1012, PIs Jimenez (VUmc), Giovannetti (VUmc), Bijlsma (AMC))
- Identification of biomarkers by whole-genome sequencing and phospho-proteomics to predict responses to high-precision cancer medicines in T-cell acute lymphoblastic leukemia (KWF Maxima 2016-10355, PIs Meijerink (Prinses Maxima), Jimenez (VUmc))
2. Identification of protein markers for non-invasive (early) detection and monitoring of cancer
To gain insight into the tumor microenvironment and identify protein biomarkers for non-invasive applications, OPL has built expertise in the measurement of extracellular proteins. Due to their extracellular localization, these proteins are more prone to end up in the blood circulation and therefore they are interesting biomarker candidates. In past years, proteomic analyses of secretomes and extracellular vesicles have been performed in multiple tumor types and have indicated that non-classical protein secretion forms an important characteristic of (aggressive) cancer cells. This makes this characteristic an interesting feature to explore for biomarker development and possibly drug targeting. Especially our published work on patient AML secretomes and exosomes (collaboration Dr. Jacqueline Cloos) that implicates transfer of RNA splice complexes has received media attention and numerous orals at meetings.
Most prominent progress in the area of non-invasive markers has been achieved with the identification and validation of protein markers for colorectal cancer screening. Over the past years, tens of candidate protein markers for screening for colorectal cancer have been identified in stool and recently validated in faecal samples of ~300 individuals (colorectal cancer patients, people harboring colorectal adenomas, and healthy controls) (De Wit et al., Annals of Internal Medicine, 2017). The most promising, validated biomarker candidates emerging from the proteomics screens are being used for further development of an antibody assay that is complementary to the immunological hemoglobin test of the national screening program for colorectal cancer in the Netherlands. This research is one of the mainstays of OPL research, showcasing OPL’s expertise (and that of their collaborating partners (former VUmc pathology, now at NKI)). It has culminated in several publications, several awarded research grants, media coverage, and patents.
Finally, proteomics of platelets is another promising non-invasive strategy for disease detection, for which proof-of-concept has already been generated in years past.
Publications
- Wojtuszkiewicz, G.J. Schuurhuis, F.L. Kessler, S. Piersma, J. Knol, T.V. Pham, G. Jansen, R.J.P. Musters, J. van Meerloo, Y.G. Assaraf, G.J.L. Kaspers, S. Zweegman, J. Cloos*, C.R. Jimenez*. Exosomes secreted by apoptosis-resistant AML blasts harbor regulatory network proteins potentially involved in antagonism of apoptosis. Mol Cell Proteomics. 2016 *Shared senior authors.
- Warmoes M, Lam SW, der Groep PV, Jaspers JE, Smolders YH, de Boer L, Pham TV, Piersma SR, Rottenberg S, Boven E, Jonkers J, van Diest PJ, Jimenez CR. Secretome proteomics reveals candidate non-invasive biomarkers of BRCA1 deficiency in breast cancer. Oncotarget. 2016 Aug 23.
- van Linde ME, van der Mijn JC, Pham TV, Knol JC, Wedekind LE, Hovinga KE, Aliaga ES, Buter J, Jimenez CR, Reijneveld JC, Verheul HM. Evaluation of potential circulating biomarkers for prediction of response to chemoradiation in patients with glioblastoma. J Neurooncol. 2016 Sep;129(2):221-30.
- Rovithi M, Lind JS, Pham TV, Voortman J, Knol JC, Verheul HM, Smit EF, Jimenez CR. Response and toxicity prediction by MALDI-TOF-MS serum peptide profiling in patients with non-small cell lung cancer. Proteomics Clin Appl. 2016 Jul;10(7):743-9.
- Knol, J.C., de Reus, I., Schelfhorst, T., Beekhof, R., de Wit, M., Piersma, S.R., Pham, T.V., Smit, E.F., Verheul, H.M.W., Jiménez. C.R. Peptide-mediated ‘miniprep’ isolation of extracellular vesicles is suitable for high-throughput proteomics. EuPA Open Proteomics Volume 11, June 2016, Pages 11–15.
- Bijnsdorp IV, Maxouri O, Kardar A, Schelfhorst T, Piersma SR, Pham TV, Vis A, van Moorselaar RJ, Jimenez CR. Feasibility of urinary extracellular vesicle proteome profiling using a robust and simple, clinically applicable isolation method. J Extracell Vesicles. 2017 Apr 28;6(1):1313091.
- De Wit, M*., Bosch, L*.,….RJA Fijneman, B Carvalho, CR. Jimenez#, GA Meijer#. Novel Stool-Based Protein Biomarkers for Improved Colorectal Cancer Screening, A Case–Control Study; *Shared first authors; #Shared senior authors. Annals of Internal Medicine, 2017 Accepted manuscript
On-going projects
- Molecular Early Detection of Colorectal Cancer (SU2C MEDOCC, Dream team leaders Meijer and Velculescu)
- Urinary extracellular vesicles and their content as novel markers for minimally invasive diagnosis and prognosis of prostate cancer (KWF/Alpe d’Huzes EMCR 2015-8022 (PIs Jenster (ErasmusMC), Jimenez (VUmc), Schalken (RadboudMC))
3. Development/ implementation of innovative proteomics and data analysis strategies
Innovation of research methodologies and analysis strategies is an on-going effort, often as a spin-off of specific questions or problems addressed in regular projects. OPL’s basic, label-free proteomics workflow is based on GeLC-MS/MS and spectral counting. This workflow for global protein identification and quantification has proved very robust and reliable, and the pertinent publication where this procedure was compared to other approaches is one of OPL’s most cited papers (Piersma et al., JPR 2010). For the analysis of spectral count data, OPL has devised a dedicated statistical test (Pham et al., Bioinformatics 2010; 2012). To speed up the gel band processing step in the GeLC workflow, we introduced the “whole gel” protocol. To enable analysis of tumor biopsies, we scaled-down the pTyr phosphoproteomics workflow. Furthermore, OPL is actively engaged in the development of algorithms that make prediction of driver kinases from phosphoproteome data possible (unpublished data). Finally a proteogenomics pipeline to enable analysis of alternative splicing was recently developed (Komor et al., Mol.Cell.Proteomics 2017).
Publications
- Piersma, SR., Knol, JC., de Reus, I., Labots, M., Sampadi, BK., Pham, TV., Ishihama, Y., Verheul, HMW., Jimenez, CR. Feasibility of label-free phosphoproteomics and application to base-line signaling of colorectal cancer cell lines. J. Proteomics 2015, doi:10.1016/j.jprot.2015.03.019.
- Van der Mijn, J. C., Labots, M., Piersma, S. R., Pham, T. V, Knol, J. C., Broxterman, H. J., Verheul, HM., Jiménez, CR. Evaluation of different phospho-tyrosine antibodies for label-free phosphoproteomics. J. Proteomics 2015, doi:10.1016/ j.jprot.2015.04.006.
- Labots M, van der Mijn JC, Beekhof R, Piersma SR, de Goeij-de Haas RR, Pham TV, Knol JC, Dekker H, van Grieken NCT, Verheul HMW, Jimenez CR. Phosphotyrosine-based-phosphoproteomics scaled-down to biopsy level for analysis of individual tumor biology and treatment selection. J Proteomics. 2017 Apr 23. pii: S1874-3919(17)30140-9.
- Komor MA, Pham TV, Hiemstra AC, Piersma SR, Bolijn AS, Schelfhorst T,Delis-van Diemen PM, Tijssen M, Han SK, Sebra RP, Ashby M, Meijer GA, Jimenez CR, Fijneman RJA. Identification of differentially expressed splice variants by the proteogenomic pipeline SPLICIFY. Molecular Cellular Prot. 2017 Accepted Manuscript.
On-going projects
- Kinome pathway activity assay by targeted mass spectrometry-based phosphoproteomics to enable personalized treatment of cancer with kinase inhibitors (CCA 2016-2017, PIs Jimenez, Grieken, Verheul)
- Next-generation proteomics to create digital cancer proteome maps for translational research and personalized medicine (NWO Middelgroot, PIs Jimenez, Verheul)
Training & Dissemination
Training in proteomic analysis strategies and dissemination of strategies developed within the OPL are important aspects of our mission. To this end, the OPL is continuously working with and mentoring postdoctoral scholars, graduate and undergraduate students.
Additionally, one master-level course (Biomedical Proteomics) is coordinated by the OPL. This course is open to other researchers, and every year also a number of PhD students and collaborators attend. Furthermore, we teach several classes in the course ‘Bioinformatics for Translational Medicine (B4TM)’, in the VUmc master oncology as well as in bachelor courses.
The OPL publishes extensively in peer-reviewed journals (see publications) and also presents its research at a range of international conferences and forums.
Through this website we also make available protocols (eg., tissue lysate preparation, gel staining, in-gel digest protocols) and software tools (beta-binomial test) used by the OPL.
Progress Reports
The OPL progress reports covers the cancer proteomics research of the OncoProteomics Laboratory (OPL) and collaborators, as well as proteomics activities performed in the context of our function as Enabling Technology Center / Core Facility. They harbor information on our mass spectrometry infrastructure and proteomics expertise, output, academic activities and includes summaries of running core and collaborative projects (detailed in appendix 1).